Selected ATcT [1, 2] enthalpy of formation based on version 1.176 of the Thermochemical Network [3]
This version of ATcT results[3] was generated by additional expansion of version 1.172 to include species related to Criegee intermediates that are involved in several ongoing studies[4].Allyl anion
2D Image:
![C=C[CH2-]](../images/501.png)
| ΔfH°(0 K) | ΔfH°(298.15 K) | Uncertainty | Units |
|---|---|---|---|
| 134.00 | 122.99 | ± 0.21 | kJ/mol |
3D Image of [CH2CHCH2]- (g)
Top contributors to the provenance of ΔfH° of [CH2CHCH2]- (g)
The 20 contributors listed below account only for 58.1% of the provenance of ΔfH° of [CH2CHCH2]- (g).A total of 445 contributors would be needed to account for 90% of the provenance.
Please note: The list is limited to 20 most important contributors or, if less, a number sufficient to account for 90% of the provenance. The Reference acts as a further link to the relevant references and notes for the measurement. The Measured Quantity is normaly given in the original units; in cases where we have reinterpreted the original measurement, the listed value may differ from that given by the authors. The quoted uncertainty is the a priori uncertainty used as input when constructing the initial Thermochemical Network, and corresponds either to the value proposed by the original authors or to our estimate; if an additional multiplier is given in parentheses immediately after the prior uncertainty, it corresponds to the factor by which the prior uncertainty needed to be multiplied during the ATcT analysis in order to make that particular measurement consistent with the prevailing knowledge contained in the Thermochemical Network.
Top 10 species with enthalpies of formation correlated to the ΔfH° of [CH2CHCH2]- (g)
The correlation coefficient is a number from -1 to 1, with 1 representing perfectly correlated species, -1 representing perfectly anti-correlated species, and 0 representing perfectly uncorrelated species.
| Correlation Coefficent (%) | Species Name | Formula | Image | ΔfH°(0 K) | ΔfH°(298.15 K) | Uncertainty | Units | Relative Molecular Mass | ATcT ID |
|---|---|---|---|---|---|---|---|---|---|
| 81.3 | Propene | CH3CHCH2 (g) | 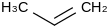 | 34.89 | 20.06 | ± 0.18 | kJ/mol | 42.0797 ± 0.0024 | 115-07-1*0 |
| 81.3 | Propylene cation | [CH3CHCH2]+ (g) | ![CC=[CH2+]](../images/1625.png) | 975.18 | 961.65 | ± 0.18 | kJ/mol | 42.0792 ± 0.0024 | 34504-10-4*0 |
| 48.8 | Propane | CH3CH2CH3 (g) |  | -82.73 | -105.01 | ± 0.15 | kJ/mol | 44.0956 ± 0.0025 | 74-98-6*0 |
| 38.2 | n-Butane | CH3CH2CH2CH3 (g) |  | -98.25 | -125.56 | ± 0.18 | kJ/mol | 58.1222 ± 0.0033 | 106-97-8*0 |
| 37.7 | Ethane | CH3CH3 (g) |  | -68.40 | -84.03 | ± 0.12 | kJ/mol | 30.0690 ± 0.0017 | 74-84-0*0 |
| 35.4 | Ethylene | CH2CH2 (g) |  | 60.89 | 52.38 | ± 0.11 | kJ/mol | 28.0532 ± 0.0016 | 74-85-1*0 |
| 35.4 | Ethylene cation | [CH2CH2]+ (g) | ![C=[CH2+]](../images/1152.png) | 1075.21 | 1067.99 | ± 0.11 | kJ/mol | 28.0526 ± 0.0016 | 34470-02-5*0 |
| 31.3 | iso-Propylium | [CH3CHCH3]+ (g) | ![C[CH+]C](../images/313.png) | 822.97 | 805.89 | ± 0.23 | kJ/mol | 43.0871 ± 0.0024 | 19252-53-0*0 |
| 30.1 | Carbonic acid | C(O)(OH)2 (aq, undissoc) | 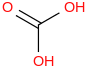 | -698.673 | ± 0.028 | kJ/mol | 62.0248 ± 0.0012 | 463-79-6*1000 | |
| 29.5 | Propyne | CH3CCH (g) |  | 192.70 | 185.55 | ± 0.20 | kJ/mol | 40.0639 ± 0.0024 | 74-99-7*0 |
Most Influential reactions involving [CH2CHCH2]- (g)
Please note: The list, which is based on a hat (projection) matrix analysis, is limited to no more than 20 largest influences.| 1 |
B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszewski, S. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff, and A. F. Wagner, Introduction to Active Thermochemical Tables: Several "Key" Enthalpies of Formation Revisited. J. Phys. Chem. A 108, 9979-9997 (2004) [DOI: 10.1021/jp047912y] |
|
| 2 |
B. Ruscic, R. E. Pinzon, G. von Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, D. Montoya, and A. F. Wagner, Active Thermochemical Tables: Thermochemistry for the 21st Century. J. Phys. Conf. Ser. 16, 561-570 (2005) [DOI: 10.1088/1742-6596/16/1/078] |
|
| 3 |
B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.176 of the Thermochemical Network (2024); available at ATcT.anl.gov |
|
| 4 |
T. L. Nguyen et al, ongoing studies (2024) |
|
| 5 |
B. Ruscic, Uncertainty Quantification in Thermochemistry, Benchmarking Electronic Structure Computations, and Active Thermochemical Tables. Int. J. Quantum Chem. 114, 1097-1101 (2014) [DOI: 10.1002/qua.24605] |
|
| 6 |
B. Ruscic and D. H. Bross, Thermochemistry Computer Aided Chem. Eng. 45, 3-114 (2019) [DOI: 10.1016/B978-0-444-64087-1.00001-2] |
Note that an uncertainty of ± 0.000 kJ/mol indicates that the estimated uncertainty is < ± 0.0005 kJ/mol.
The find function is based on the complete Species Dictionary entries for the appropriate version of the ATcT TN.
The molecule images are rendered by Indigo-depict.
The XYZ renderings are based on Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/.