loadScript ./j2s/core/package.js
loadScript ./j2s/core/corejmol.z.js
JSmol exec jmolApplet0 start applet null
Jmol JavaScript applet jmolApplet0__813262151629021__ initializing
Jmol getValue debug null
Jmol getValue logLevel null
Jmol getValue allowjavascript true
AppletRegistry.checkIn(jmolApplet0__813262151629021__)
vwrOptions:
{ "name":"jmolApplet0","allowJavaScript":true,"applet":true,"documentBase":"https://atct.anl.gov/Thermochemical%20Data/version%201.122d/species/?species_number=404","platform":"J.awtjs2d.Platform","fullName":"jmolApplet0__813262151629021__","codePath":"https://atct.anl.gov/Thermochemical%20Data/version%201.122d/species/./j2s/","display":"jmolApplet0_canvas2d","signedApplet":"true","appletReadyCallback":"Jmol._readyCallback","statusListener":"[J.appletjs.Jmol object]","syncId":"813262151629021","bgcolor":"#FFFFFF" }
setting document base to "https://atct.anl.gov/Thermochemical%20Data/version%201.122d/species/?species_number=404"
(C) 2015 Jmol Development
Jmol Version: 14.6.1_2016.08.11 2016-08-11 22:11
java.vendor: Java2Script (HTML5)
java.version: 2016-08-02 04:55:11 (JSmol/j2s)
os.name: Mozilla/5.0 AppleWebKit/537.36 (KHTML, like Gecko; compatible; ClaudeBot/1.0; +claudebot@anthropic.com)
Access: ALL
memory: 0.0/0.0
processors available: 1
useCommandThread: false
appletId:jmolApplet0 (signed)
loadScript ./j2s/core/corescript.z.js
Jmol getValue emulate null
defaults = "Jmol"
Jmol getValue boxbgcolor null
Jmol getValue bgcolor #FFFFFF
backgroundColor = "#FFFFFF"
Jmol getValue ANIMFRAMECallback null
Jmol getValue APPLETREADYCallback Jmol._readyCallback
APPLETREADYCallback = "Jmol._readyCallback"
callback set for APPLETREADYCallback Jmol._readyCallback APPLETREADY
Jmol getValue ATOMMOVEDCallback null
Jmol getValue CLICKCallback null
Jmol getValue DRAGDROPCallback null
Jmol getValue ECHOCallback null
Jmol getValue ERRORCallback null
Jmol getValue EVALCallback null
Jmol getValue HOVERCallback null
Jmol getValue IMAGECallback null
Jmol getValue LOADSTRUCTCallback null
Jmol getValue MEASURECallback null
Jmol getValue MESSAGECallback null
Jmol getValue MINIMIZATIONCallback null
Jmol getValue SERVICECallback null
Jmol getValue PICKCallback null
Jmol getValue RESIZECallback null
Jmol getValue SCRIPTCallback null
Jmol getValue SYNCCallback null
Jmol getValue STRUCTUREMODIFIEDCallback null
Jmol getValue doTranslate null
language=en_US
Jmol getValue popupMenu null
Jmol getValue script null
Jmol getValue loadInline null
Jmol getValue load null
Jmol applet jmolApplet0__813262151629021__ ready
script 1 started
zoomLarge = false
antialiasDisplay = true
FileManager.getAtomSetCollectionFromFile(../xyz/C6H5NO.xyz)
FileManager opening url https://atct.anl.gov/Thermochemical%20Data/version%201.122d/xyz/C6H5NO.xyz
The Resolver thinks Xyz
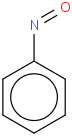
Nitrosobenzene 1A' Cs B3LYP/cc-pVTZ+d
Nitrosobenzene 1A' Cs B3LYP/6-31G(2df,p)
Time for openFile(../xyz/C6H5NO.xyz): 96 ms
reading 26 atoms
ModelSet: haveSymmetry:false haveUnitcells:false haveFractionalCoord:false
2 models in this collection. Use getProperty "modelInfo" or getProperty "auxiliaryInfo" to inspect them.
Default Van der Waals type for model set to Babel
26 atoms created
ModelSet: autobonding; use autobond=false to not generate bonds automatically
Time for creating model: 34 ms
Nitrosobenzene 1A' Cs B3LYP/cc-pVTZ+d
Nitrosobenzene 1A' Cs B3LYP/6-31G(2df,p)
2 models
loadScript ./j2s/J/thread/SpinThread.js
Script completed
Jmol script terminated
spinFPS is set too fast (30) -- can't keep up!

![c1cccc[c+]1](../images/138.png)
![c1cccc[c+]1](../images/931.png)
[O-]](../images/563.png)
[O-]](../images/1107.png)
![c1ccc(cc1)[I+]](../images/1104.png)
![c1cccc[c]1](../images/98.png)
![c1ccc(cc1)[Br+]](../images/561.png)